Currently, the Crandall Lab is examining ways to use the underlying OpenTree taxonomy to gather metadata, associate it with nodes and tips in our synthetic trees, and apply it to evolutionary studies. Below we discuss them in the context of ongoing projects in the lab.
Curated taxonomy
One of the major outcomes of the OpenTree project is the underlying taxonomy. A curated taxonomy allows us to search and align names across independent databases to pull out additional information to associate with node and tip names. The Crandall Lab taxonomy curation started with the freshwater crayfish, which are in the Infraorder Astacidea and includes 711 species spread among 7 families. This was a great group to start with because of the limited number of species and there are only a few active systematists revising the alpha-taxonomy, which makes the literature less dense and easier to work with. Initially, our investigations deemed the taxonomy very accurate, but the main issue we had to contend with was spelling errors attributed to depositing sequences into GenBank. In all, we identified and removed 10 misspelled taxa. Although that seems small, it was a great warm-up for two larger groups we are now working on, the Decapoda (crabs, shrimps, lobsters) which includes ~15,000 species and the Hemiptera (true bugs) which includes ~ 50,000-80,000 species.
Using a curated taxonomy to obtain additional data
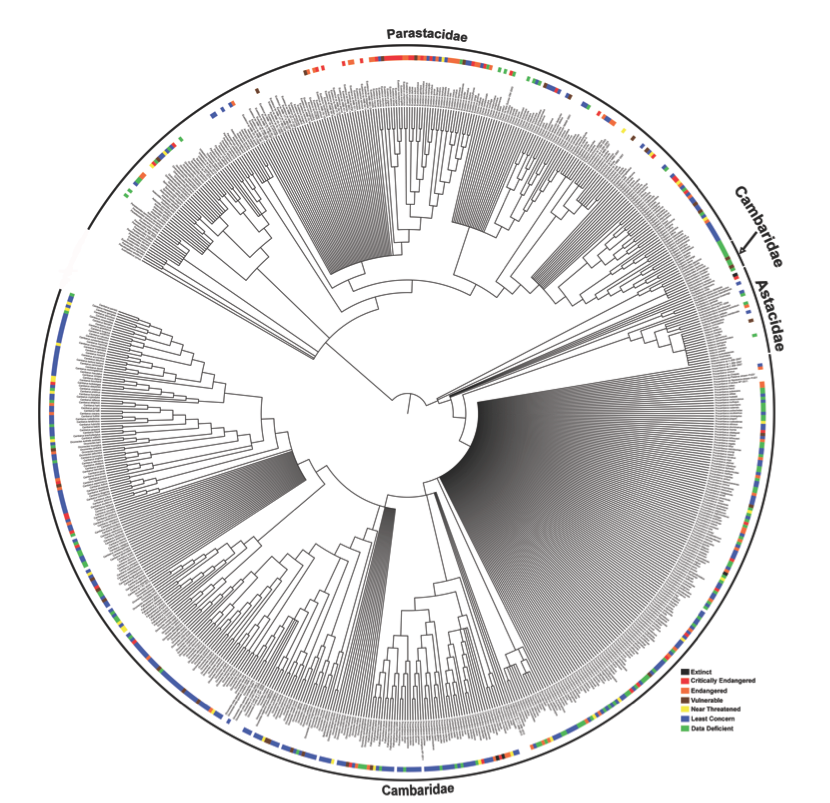
As mentioned above, once the names have been curated we can use them to search across databases. This has been extremely useful in obtaining additional metadata to associate with our synthetic trees. For example, the Crandall Lab recently published a synthetic tree of the crayfish (Fig. 1), which included IUCN Red List values plotted for those taxa with assigned values (Owen et al. 2015, Richman et al. 2015). This is only feasible because we are able to search across IUCN Red Listed crayfish species using the OpenTree curated taxonomy names.
Other applications of using a curated taxonomy to obtain metadata include searching across GenBank to identify whether a particular taxon or rank has molecular data associated with it. This is useful for determining sampling strategies for new and continuing studies. For example, using the OpenTree taxonomy to search GenBank for Hemiptera families and genera, we found a wealth of sequence data has been generated for most of the higher taxa. The most diverse suborder within Hemiptera is Heteroptera and our query of names against NCBI GenBank suggests 70 of the 83 described families within Heteroptera have sequence data for one or more of the traditional eight molecular loci used in Hemiptera systematics (Fig. 2A). As for the Hemiptera genera identified in GenBank, we are currently validating the numbers in Fig. 2B because Hemiptera alpha-taxonomy is very active because many species are vectors for human pathogens and agricultural pests (e.g., kissing bug, aphids, psyllids, etc.).
In addition to searching GenBank, we are currently associating geographic, morphological, and ecological metadata to our curated names through GBIF and EOL TraitBank. We believe the curated OpenTree taxonomies of these groups and the accumulation of metadata for taxa will surely add a new dimension to our evolutionary studies and allow us to expand the scope the questions we can answer.

Figure 1 Synthetic tree of crayfish with 20 source trees. Family names noted on the edge of the synthetic tree. Paraphyly of Cambaridae is not novel and needs to be addressed in a morphological revision. Color blocks note the IUCN Redlist value.

Figure 2 Histograms depicting number of sequences found on GenBank given OTT names. 2A) Hemiptera families within suborders with nucleotide sequence data on NCBI GenBank. 2B) Hemiptera genera within suborders with nucleotide sequence data on NCBI GenBank.
Keith Crandall is a professor and director of the Computational Biology Institute at George Washington University.
Chris Owen is a post-doctoral researcher for the AVAToL grant.
Literature
Owen, C. L., Bracken-Grissom, H., Stern, D., & Crandall, K. A. (2015). A synthetic phylogeny of freshwater crayfish: insights for conservation.Philosophical Transactions of the Royal Society of London B: Biological Sciences, 370(1662), 20140009.
Richman, N. I., Böhm, M., Adams, S. B., Alvarez, F., Bergey, E. A., Bunn, J. J., … & Collen, B. (2015). Multiple drivers of decline in the global status of freshwater crayfish (Decapoda: Astacidea). Philosophical Transactions of the Royal Society B: Biological Sciences, 370(1662), 20140060.